
Pathological mechanisms in sphingolipidoses
Sphingolipidoses are typically caused by pathogenic variants in genes encoding lysosomal enzymes responsible for degrading sphingolipids. Most of the genes that underlie sphingolipidoses are also implicated in LBD. Sphingolipid storage disorders often affect very young children, leading to severe neurodegeneration and early mortality. As the causative gene is known for all of these disorders, most research either focuses on metabolic pathways associated with the deficient enzyme or tries to develop gene therapies to correct the deficient enzyme. We uniquely approach these disorders like we do any other neurodegenerative disorder, utilising neuropathology (including discovery proteomics) to agnostically identify implicated biological pathways, then screen compounds that act upon the implicated pathways in cell model systems to determine whether phenotypes can be recovered with re-purposed drugs.
Metachromatic leukodystrophy
MLD is a rare lysosomal storage disorder caused by bi-allelic pathogenic variants in the ARSA gene, which encodes the lysosomal enzyme arylsulphatase A. Dysfunction of arylsulphatase A leads to the accumulation of its substrate, sulphatide, and widespread demyelination. We have also noted inflammation and activation of the nutrient-sensing mTOR pathway in MLD. MLD usually onsets in infancy or the juvenile period, but can also onset in adults. It has a progressive and unrelenting course and, tragically, leads to early mortality.
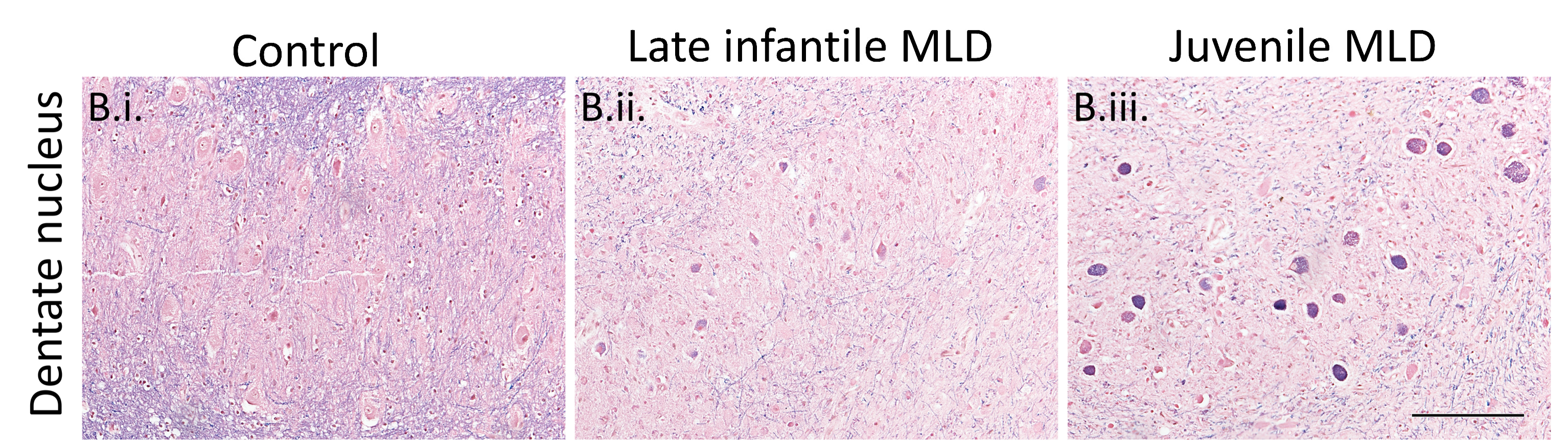 Figure 1: Luxol fast blue staining in MLD and control dentate nucleus. The loss of blue staining is apparent in MLD cases, indicating demyelination, alongside intraneuronal accumulation that is especially marked in older juvenile cases.
Figure 1: Luxol fast blue staining in MLD and control dentate nucleus. The loss of blue staining is apparent in MLD cases, indicating demyelination, alongside intraneuronal accumulation that is especially marked in older juvenile cases.
Krabbe disease
Krabbe disease is also a rare lysosomal storage disorder, but it is caused by bi-allelic loss-of-function variants in the GALC gene. As with MLD, this enocdes a lysosomal enzyme, in this case galactosylceramidase, and leads to the accumulation of a de-acylated form of its substrate, galactosylsphingosine/psychosine. As with MLD, this leads to widespread demyelination and the formation of characteristic multi-nucleated globoid cells, often surrounding blood vessels. Krabbe disease typically onsets in the first few months of life and, tragically, affected infants usually die within a year of being diagnosed.






